二代测序数据基础分析(一)
技术介绍
- 第二代测序技术,1990s - 2010s
第二代测序技术从原理上分为三种方法:
- Roche 454测序法
- IIIumina Solexa/Hiseq测序法
- ABI Solid测序法
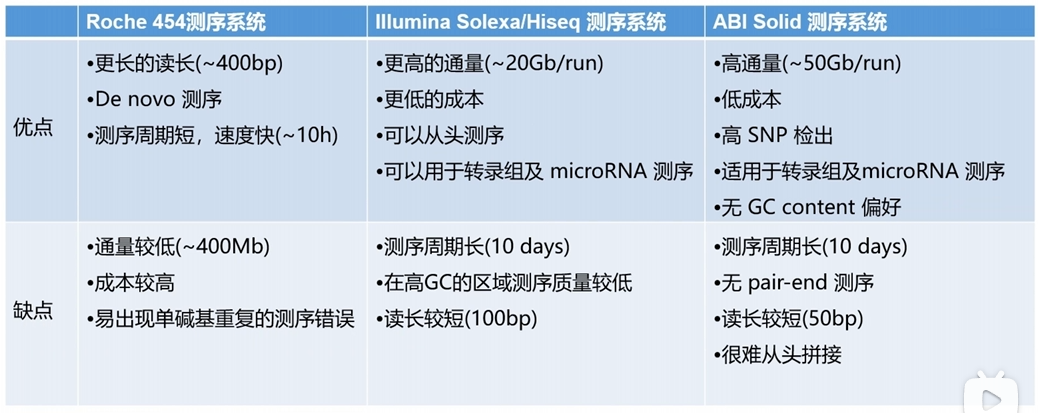
三种二代测序技术的对比:
总结
二代测序技术总体而言都有着同一种特性:
- 需荧光或化学发光物质
- 需聚合酶或连接酶
- 需购买昂贵的试剂耗材和光学系统
- 需强大的图形分析计算能力
WES分析流程
WES分析流程分为5个步骤:
- WES分析流程
- 数据质量控制
- 序列比对分析
- 变异检测
- 变异注释
一. WES分析流程
原始测序数据——数据质量控制——序列比对——变异检测——变异注释——完成
二. 数据质量控制
数据质控标准:
- 去掉reads中的接头
- 去除低质量(BP<20)碱基的reads
- 去除序列头尾的N碱基
- 去除头尾N碱基后若剩余reads长度小于40bp(双端),则丢弃该对序列
原始数据下载——sratoolkit软件
1 | 安装 |
基本参数:
fastq-dump [options] [path]
- –gzip: 使用gzip压缩输出文件
- –split-3: 设置文件拆分,如果是双端的序列,则拆分为*_1.fastq和*_2.fastq
- -O: 设置输出文件路径
数据质量控制常用的软件是FASTQC,在这里不再赘述,详情请看https://www.jinhenghaoblog.top/posts/20250828165237.html
序列比对分析
进行序列比对分析所用到的软件为BWA:
1 | 下载参考数据 |
基本参数:
bwa mem [options] [idxbase] [in1.fq] [in2.fq]
- idxbase: 指定比对参考基因组序列(建好索引)
- in1.fq、in2.fq:输入fastq文件
- -t : 设置线程,这里设置的是5,具体看计算机配置
SAM文件格式转换及排序
1 | sam转bam |
基本参数:
samtools sort [options] [in.bam] [out.prefix]
- -n: 根据reads名字排序
- -o: 输出文件
- -@: 设置线程,这里设置的是5,具体看计算机配置
标记/删除PCR重复的reads——GATK
1 | 建立输出文件夹 |
基本参数:
gatk MarkDuplicates -I [INPUT] -O [OUTPUT] -M [METRICS]
- MarkDuplicates: 鉴定重复的reads,是GATK中的一个工具
- -I : 输入文件
- -O : 输出文件
- -M: 复制度量写入的文件
变异检测
变异检测——标记flagstat
1 | gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" FixMateInformation -I test_marked.bam -O test_marked_fixed.bam -SO coordinate 1>test_log.fix 2>&1 |
基本参数:
gatk FixMateInformation -I [INPUT] -O [OUTPUT] -M [METRICS]
- FixMateInformation : 是GATK中的一个工具
- -I:输入文件
- -O:输出文件
- -SO: 输出文件的排序方式,这里选择的是coordinate
变异检测——GATK找变异
1 | 找变异 |
基本参数:
gatk BaseRecalibrator -I [INPUT] -O [OUTPUT] -M [METRICS]
- –known-sites: 已知的多态位点数据库(可以指定多个)
- -R: 参考序列文件
- -I:输入文件
- -O:输出文件
变异检测——矫正bam并生成VCF
1 | ————————矫正bam |
基本参数:
gatk HaplotypeCaller -i [INPUT] -O [OUTPUT] -M [METRICS]
- -R:参考序列文件
- -I:输入文件
- –dbsnp:dbSNP文件
- -O:输出文件
变异注释
变异注释使用的软件为Annovar软件
Annovar软件可实现三种不同的注释方式:
- 确定SNP或CNV是否导致蛋白质编码变化和确定受影响的氨基酸
- 识别特定基因组区域的变异
- 鉴定特定数据库中记录的变异
Annovar——下载数据库
1 | perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar refGene humandb/ |
基本参数:
Perl annotate_variation.pl [options]
- -buildver: 基因组对应版本,这里是hg19
- -webfrom annovar:指定从annovar库里下载,若annovar库中没有,则会从UCSC中下载
- refGene:数据库名称
- humandb/:数据库存储路径
Annovar——变异注释
1 | 输入文件格式转换 |
基本参数:
Perl table_annovar.pl [options]
- -remove: 删除所有的临时文件
- -geneanno:指定注释方法。这里表示gene-based,-regionanno表示region-based,–filer表示filter-based;
- -buildver: 指定参考基因组