从0开始做分子对接——基于Windows系统的Autodock对接教程
总结
本文介绍了在 Windows 环境下使用 AutoDock4 进行蛋白-小分子分子对接的完整流程。首先说明了AutoDock4 与 MGLTools 的安装方法,并利用 Pymol 对受体进行去水、加氢等预处理。随后,通过 AutoDockTools 对受体与配体文件计算电荷、设置扭转键并生成 pdbqt 文件。接着,设置 Grid Box 实现盲对接区域定义,生成并运行 GPF 与 DPF 文件,完成对接计算。最后,通过 Analyze 模块查看结合能结果,并用 OpenBabel 将结果转换为 pdb 文件在 Pymol 中可视化。整体流程清晰、步骤完整,为初学者开展分子对接实验提供了实用参考。
Autodock4安装
在进行简单的蛋白——小分子对接之前,需要对必要的前置软件进行安装,首先安装Autodock4。
其官网为:https://autodock.scripps.edu/download-autodock4/
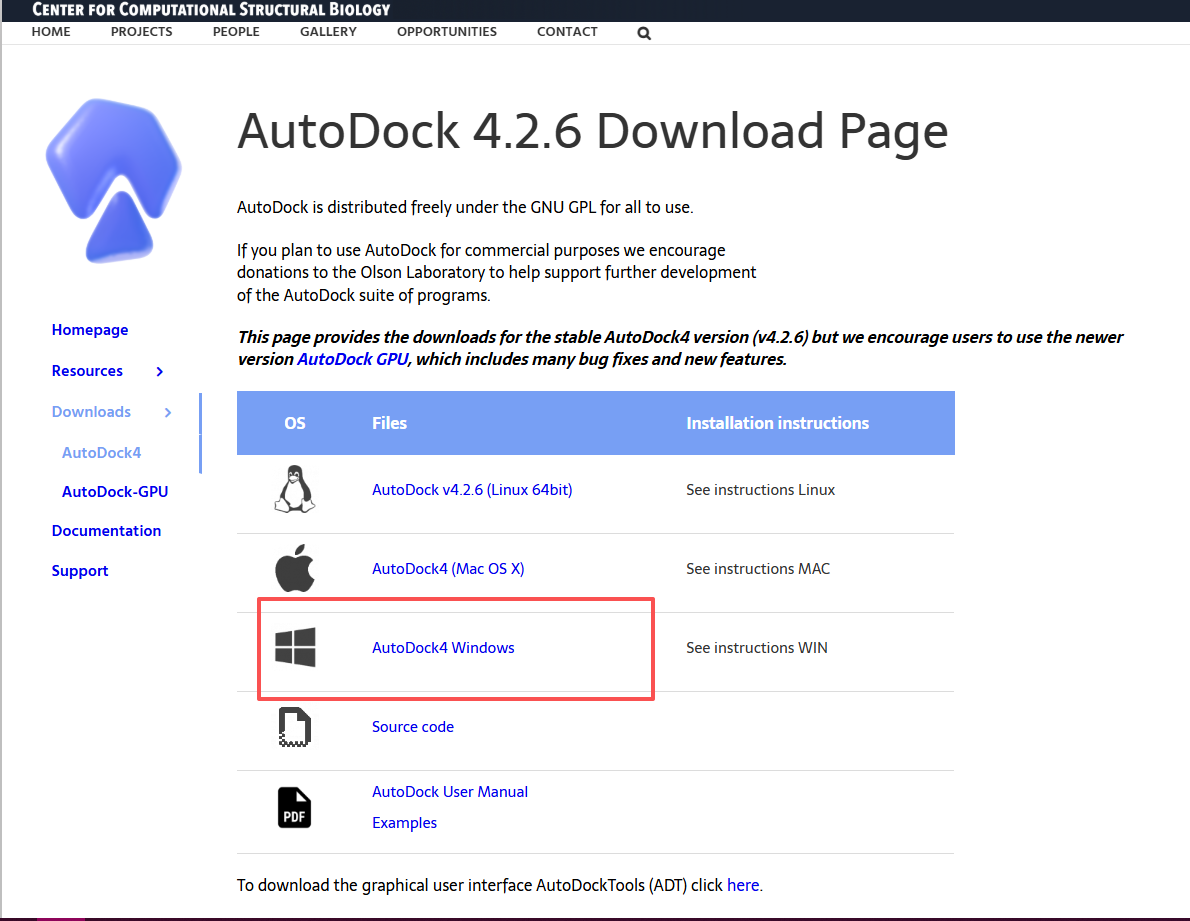
先点击Windows然后下载
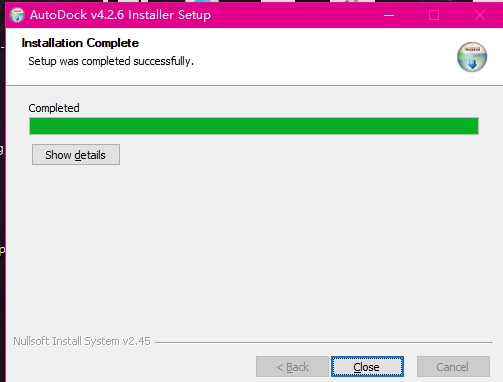
选中一个文件夹(建议新建文件夹放进去,不要用C盘),点击安装即可
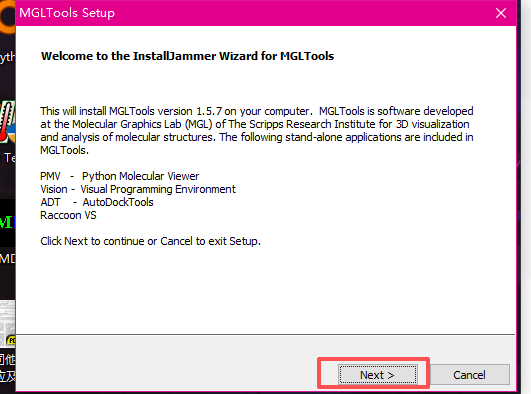
然后安装Mgltools
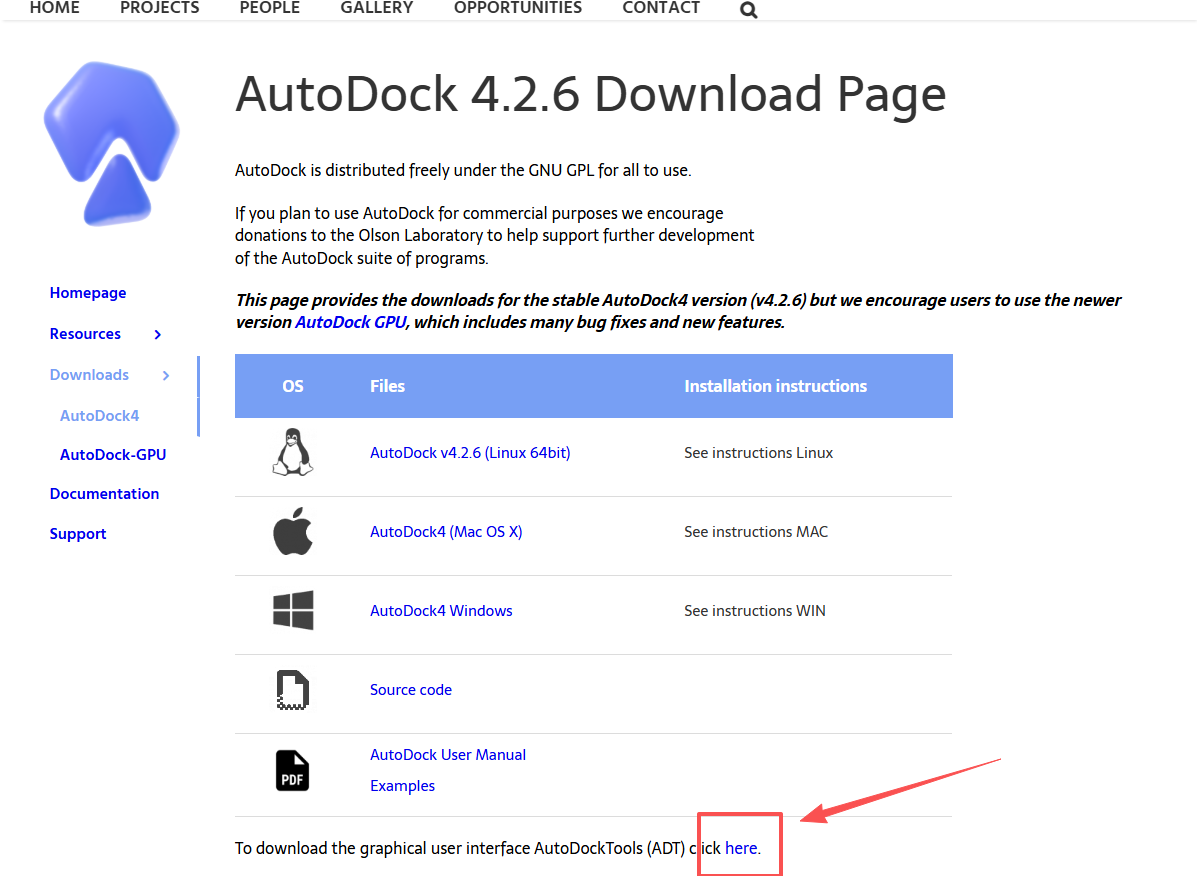
在这里我们需要选择带图形界面的版本
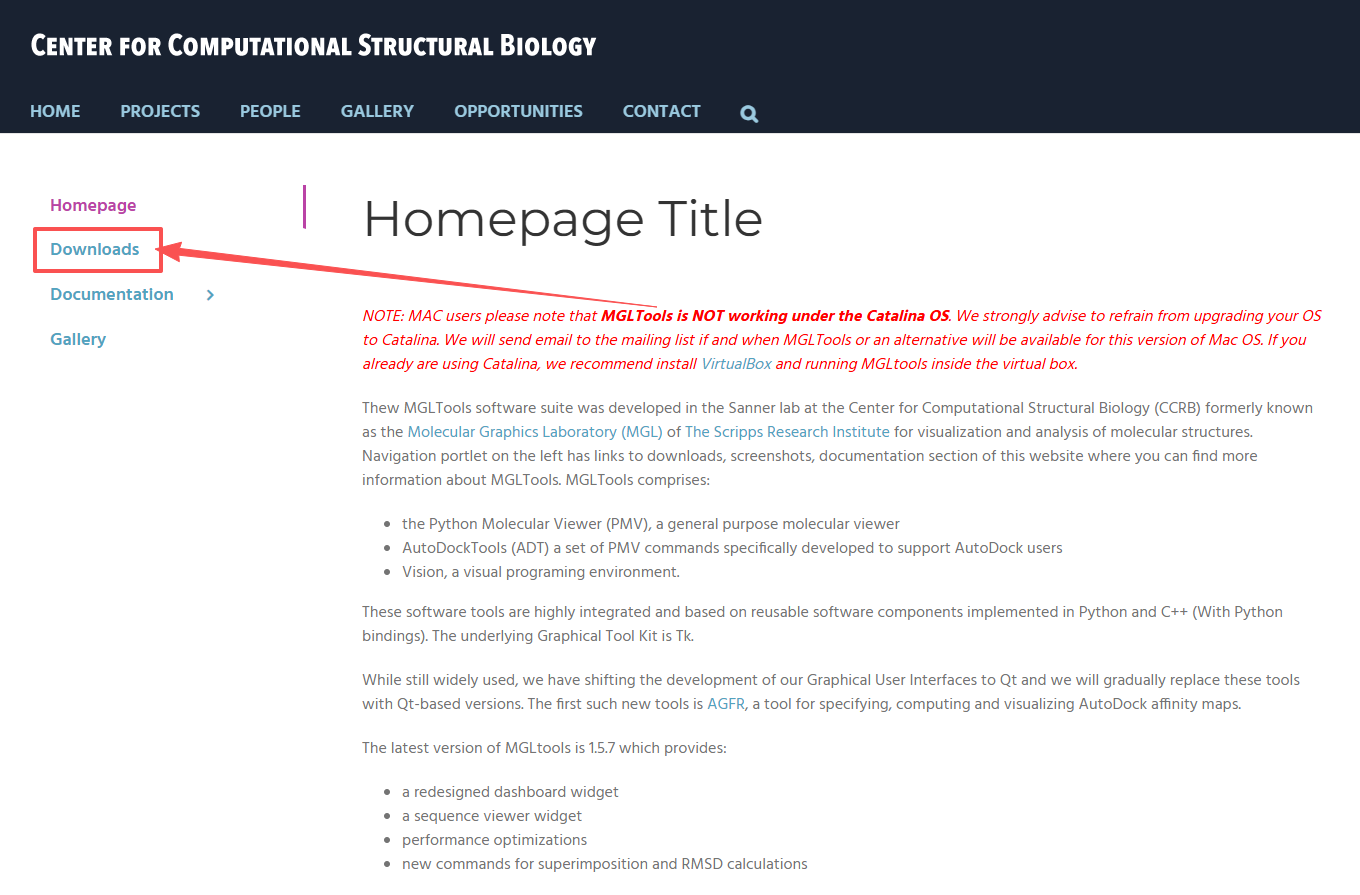
选择Downloads
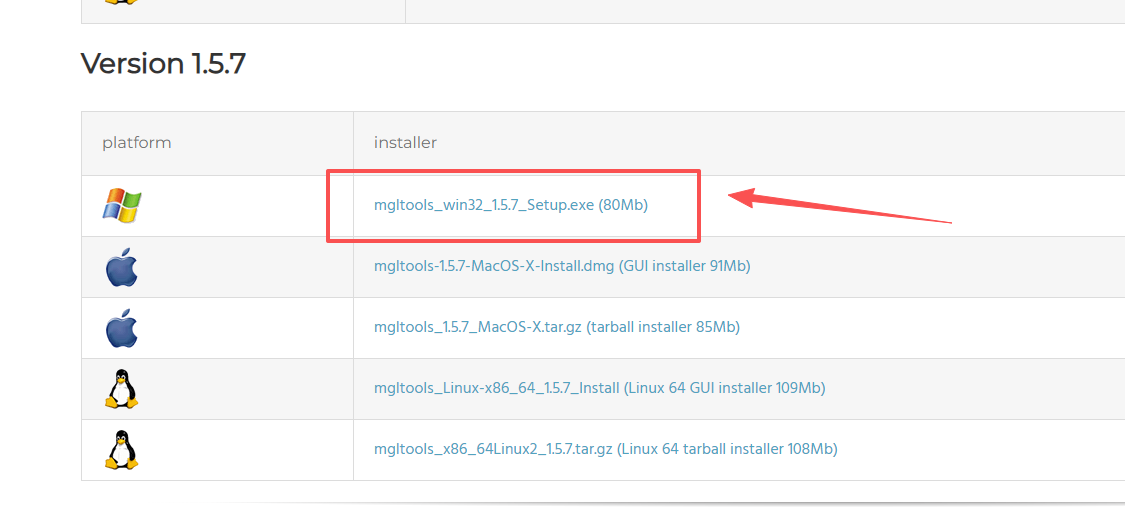
往下拉,选择mgltools_win32的版本,点击进行下载
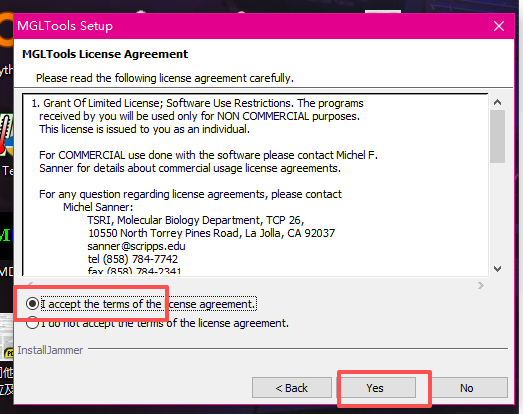
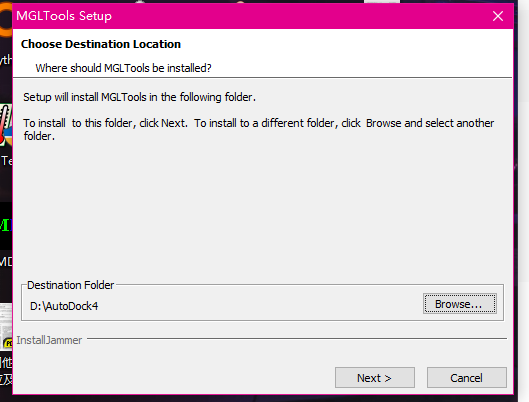
选择D盘,新建文件夹,选择,再点击Next
这样就表示安装完成
进入安装的主目录,可看见安装的文件如下:
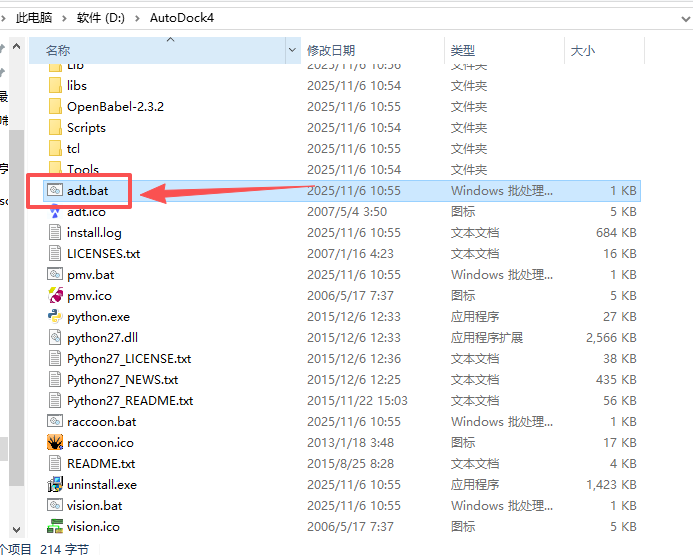
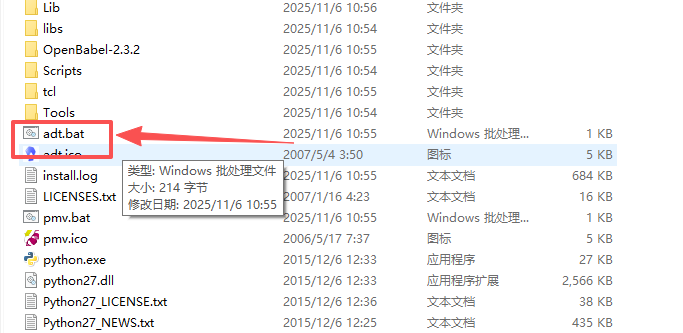
这个adt.bat就是我们所要运行的对接软件,点击出现该页面
表示AutoDock4完全安装完毕。
Pymol的安装
这里详细请看我另一篇文章:windows 下基于 anaconda 的开源版 pymol 的安装
https://www.jinhenghaoblog.top/posts/20250815202033.html
使用AutoDock进行分子对接
受体配体的文件准备
在确定对接的受体与配体后,将两者放入同一个文件夹中
如图:
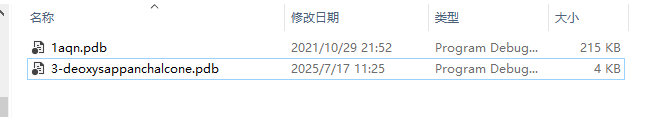
对受体来说,在做对接之前往往需要进行去水加氢的工作,因此使用Pymol打开1aqn.pdb文件
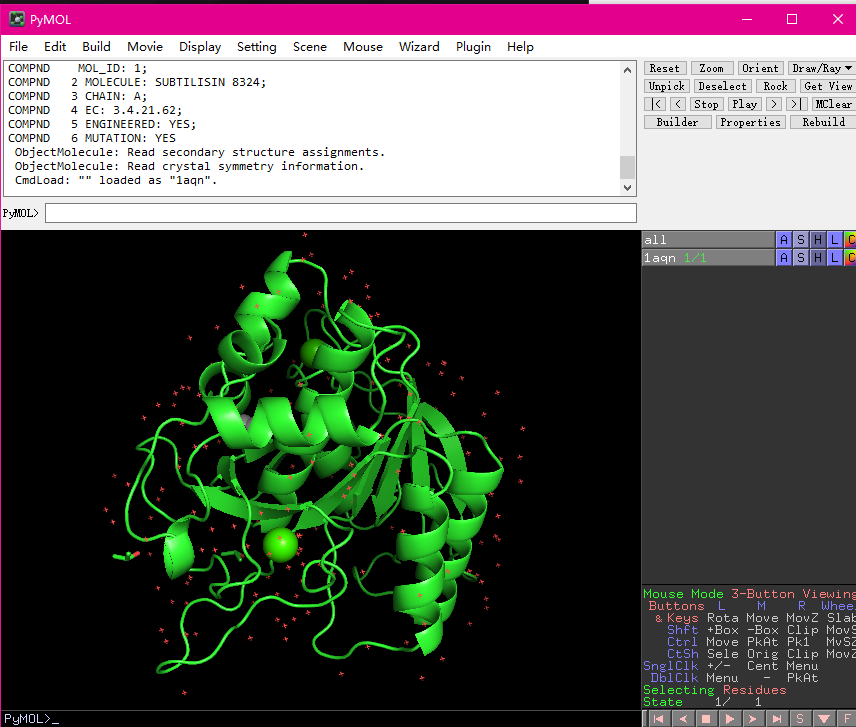
点击Display——Sequence
会看到蛋白质的序列
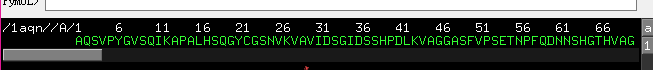
往后面拉
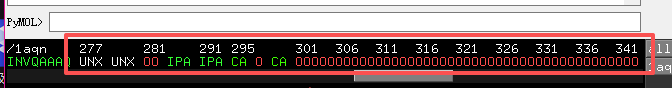
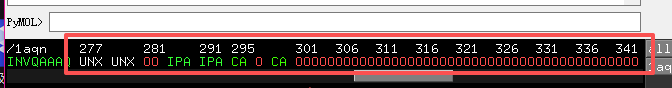
将这些小分子和红色OO全部选中
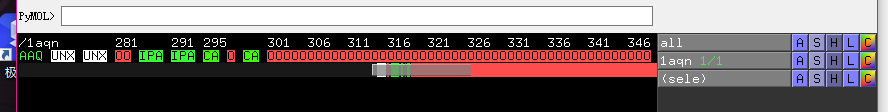
点击sele的A,点击remove atoms进行原子的删除
删除后如图所示
现在就完成了去水的工作
接下来加氢也是同样的步骤,选择点击sele的A,选择hydrogens——add进行加氢
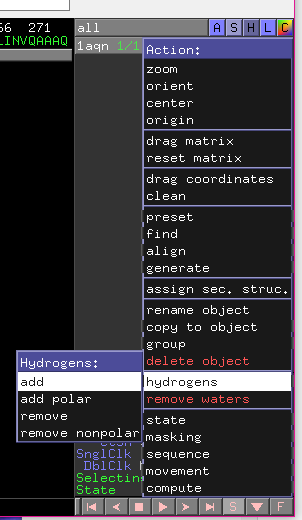
点击完毕即可
导出pdb文件,点击File——Export Molecule..
点击Save..
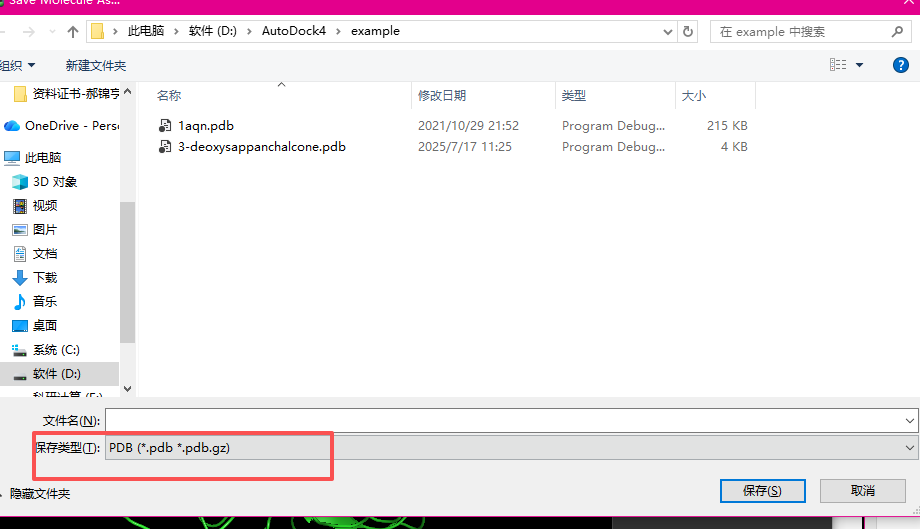
保存类型为PDB,文件名自己设置
点击保存即可
目前文件夹有这3个文件
受体文件的处理
在一切弄好之后,我们打开adt.bat,启动Autodock4
将pymol处理好的蛋白文件直接拖入AutoDock4窗口
拖入窗口后,对受体文件进行计算电荷的操作
Edit——Charges——Compute Gasteiger
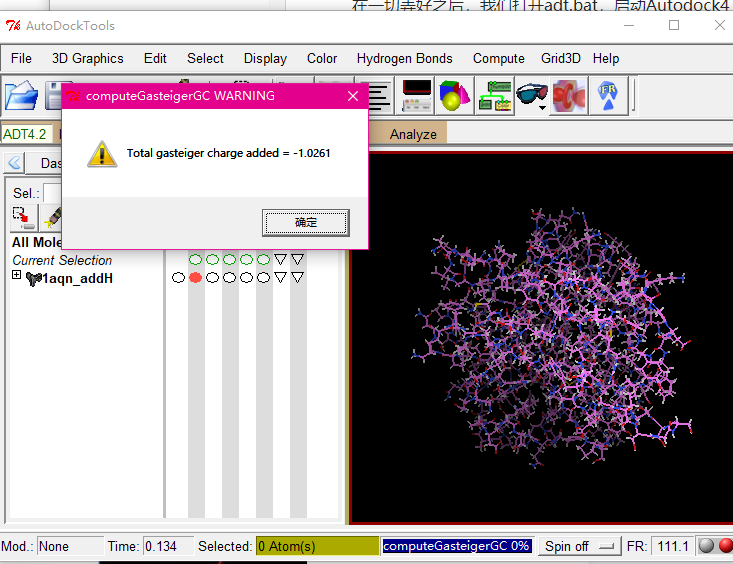
这样就表示电荷计算完成
计算完电荷后,添加原子类型
也就是:Edit——Atoms——Assign AD4 type
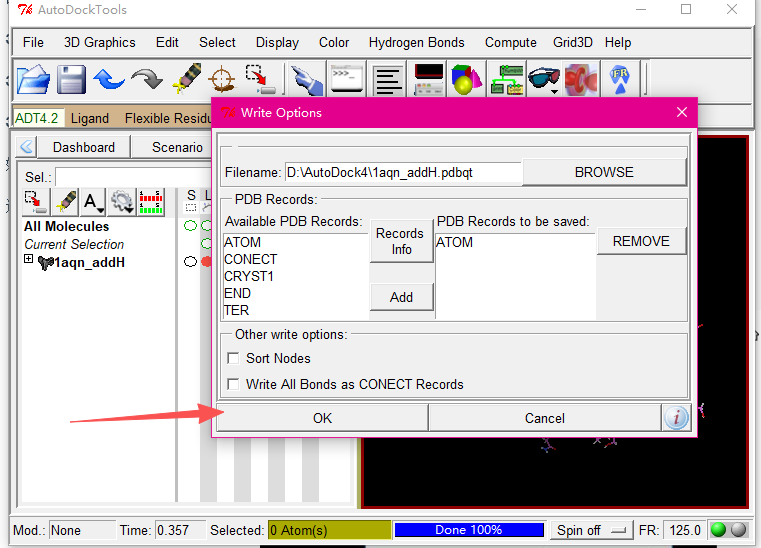
点击完成后进行保存,记得保存后缀为pdbqt文件,这个是AutoDock-Vina所需要的结构文件
也就是File——Save——Write PDBQT
配体文件的处理
将受体文件右键delete掉后,将配体文件导入至窗口中
Ligand——input——Open
点击确定
先判定配体的root,也就是
Ligand——Torsion Tree——Detect Root
然后选择配体可扭转的键
Ligand——Torsion Tree——Choose Torsions
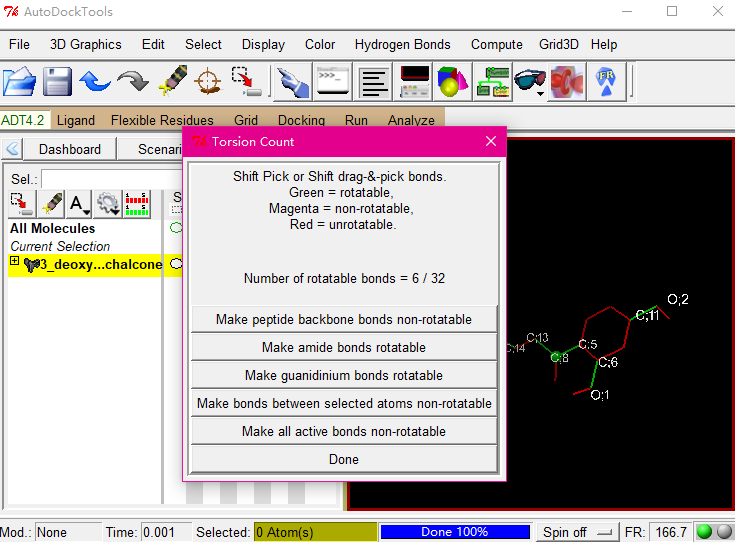
这表现分子中32个键中有6个是可以旋转的,其中红色是不可旋转的,绿色代表可旋转,紫色是不可扭转,通常为肽键,在这里我们点击Done即可
然后我们将配体保存为PDBQT文件
Ligand——Output——Save as PDBQT
设置对接区域
一般来说我们做的是盲对接,也就是将整个蛋白囊括起来,让算法探寻对接口袋,当然也有像P2rank之类的软件可以预测对接口袋,在这里我们先做的是前者。
在窗口中,我们把配体文件delete掉,然后选择
Grid——Macromocule——Open
打开受体的pdbqt文件
然后打开配体的pdbqt文件
Grid——Set Map Types——Open ligand
然后设置对接区域
Grid——Grid Box
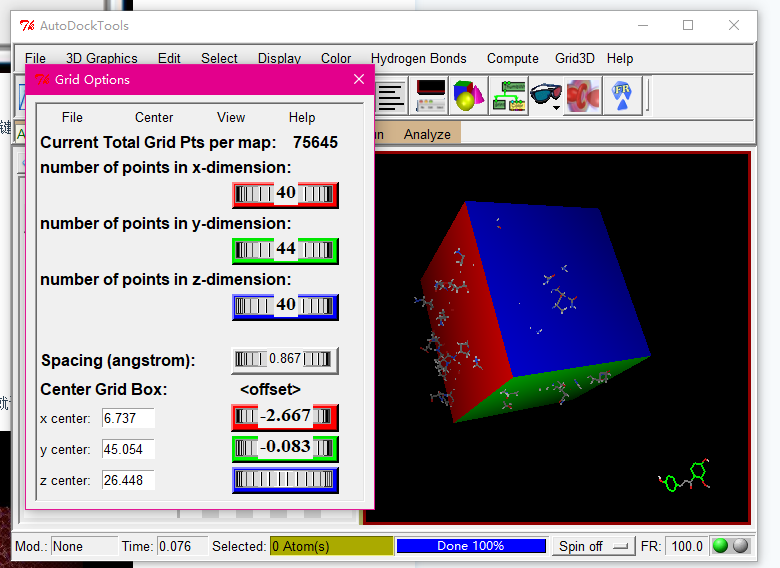
直接将整个蛋白囊括进去
调整完成后,点击Grid Options窗口里面的File——Close saving current进行保存
然后保存目前的状态为gpf文件
Grid——Output——Save GPF
保存完全后,目前文件夹的状态如下
运行AutoGrid
直接在菜单栏上点击
Run——Run Autogrid
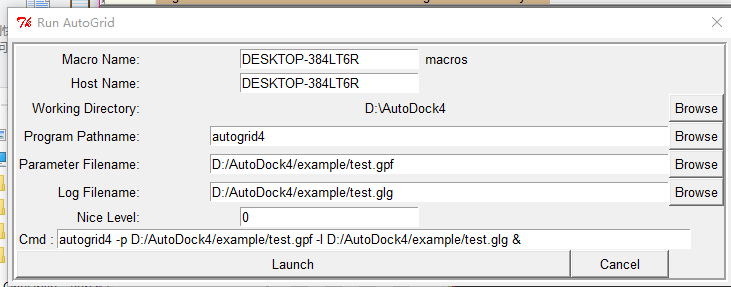
点击Launch
此时工作目录会出现若干文件
DPF文件准备
在主窗口delete两个分子,重新选择受体与配体分子
Docking——Macromocule——Set rigid macromocule,选择受体分子的PDBQT文件
Docking——Ligand——Open,选择配体分子的PDBQT文件,点击accept
然后设置对接算法
Docking——Search Parameters——Genetic Algorithm
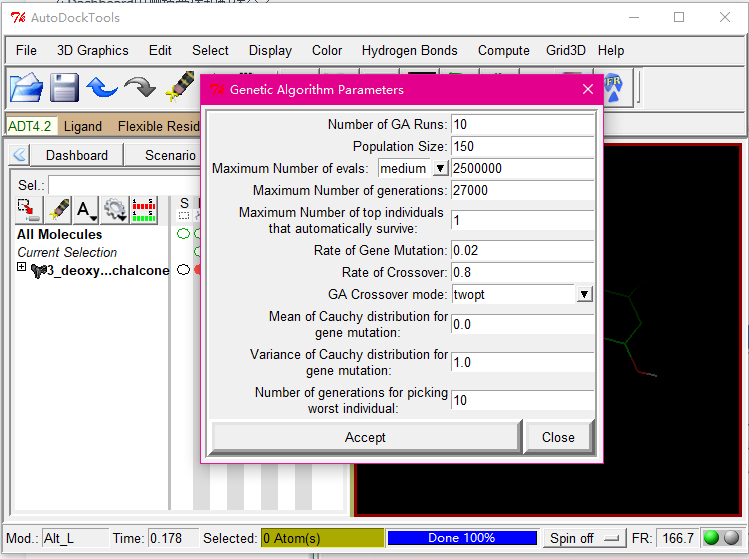
点击accept即可
然后设置对接参数
Docking——Docking Parameters
也是直接点击Accept
最后进行保存
Docking——Output——Genetic Algorithm
此时多了这个文件
运行AutoDock进行对接
直接在菜单栏上点击
Run——Run AutoDock
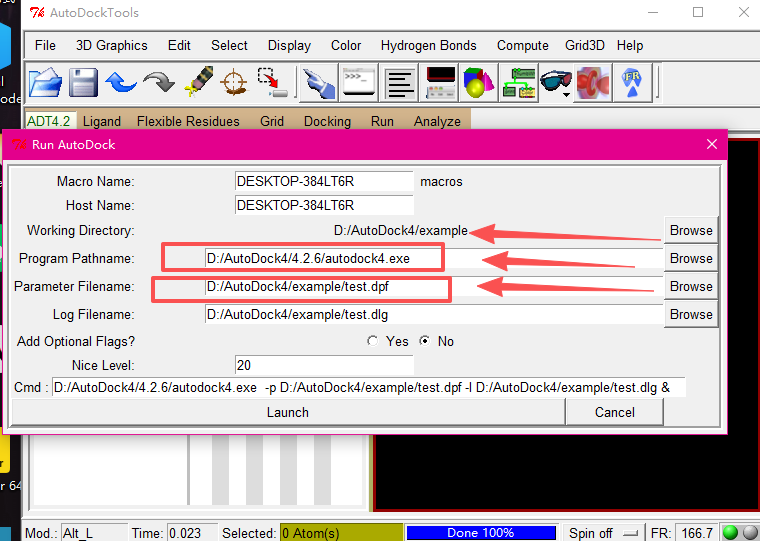
选择test.dpf还有程序文件后,点击Launch运行对接
会弹出这样的一个框框,表示加入进程中进行运行
运行对接软件需要好一会,点击任务管理器可看到Autodock在运行中
对接完毕后,目录会生成一个test.dlg文件
打开test.dlg文件,往下拉会看到本次的对接分数,分数越低,结合能越高。
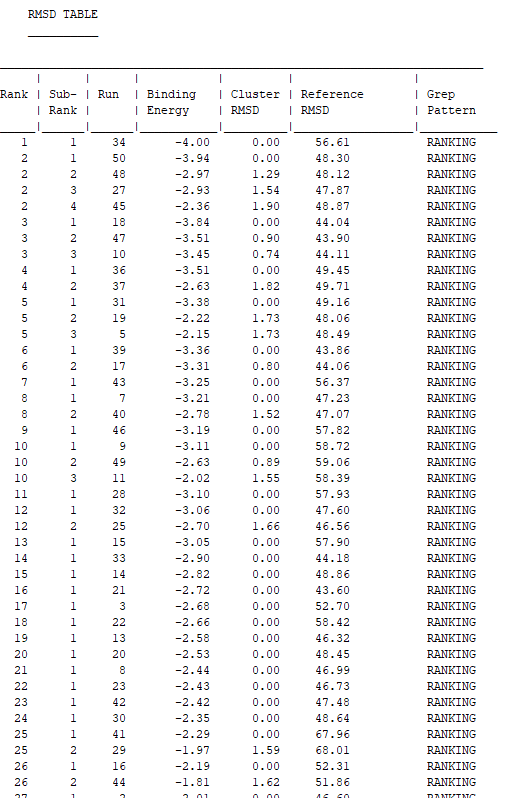
而第一列的表示是不同区域的对接分数,选择第一个就可以了
查看并导出对接结果
delete掉窗口中的小分子,加载对接结果
Analyze——Dockings——Open
打开dlg文件,加载对接小分子
再加载受体大分子
Analyze——Macromolecule——Open
点击
Analyze——Conformation——play
一个个查看对接的结果
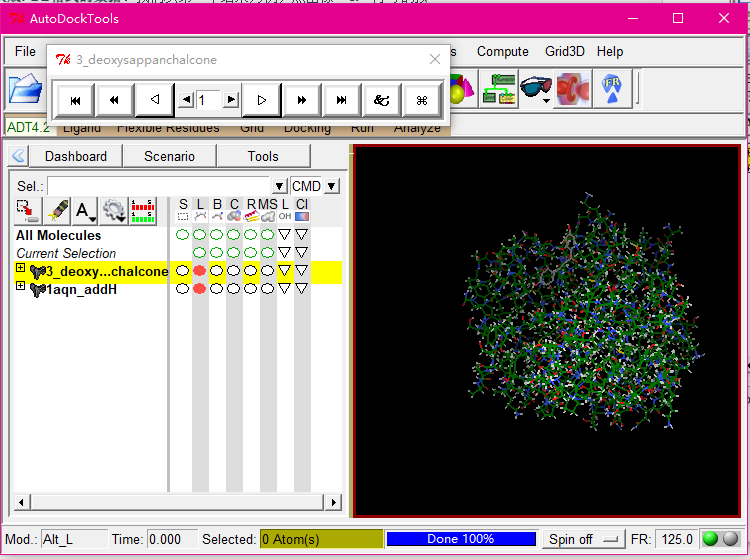
在这里我就选择第一个进行导出吧
点击
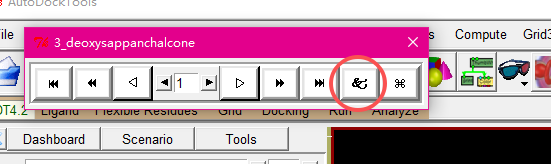
点击
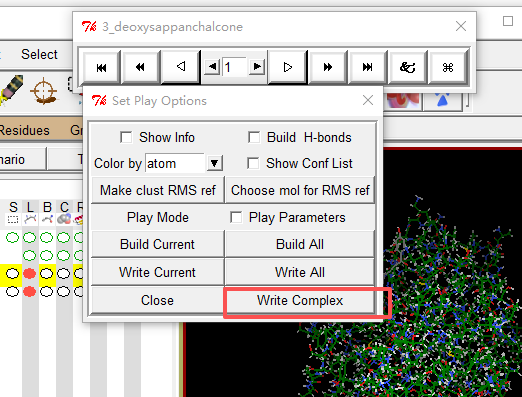
保存为pdbqt文件
如果我们想在Pymol去查看分子的话,还需要把文件转换为pdb文件才行(当然你也可以直接把pdbqt导入到pymol里面进行查看),这就需要Openbabel软件,不过随着mgltools的安装,Openbabel也会一并安装在里面
在主目录的
点击
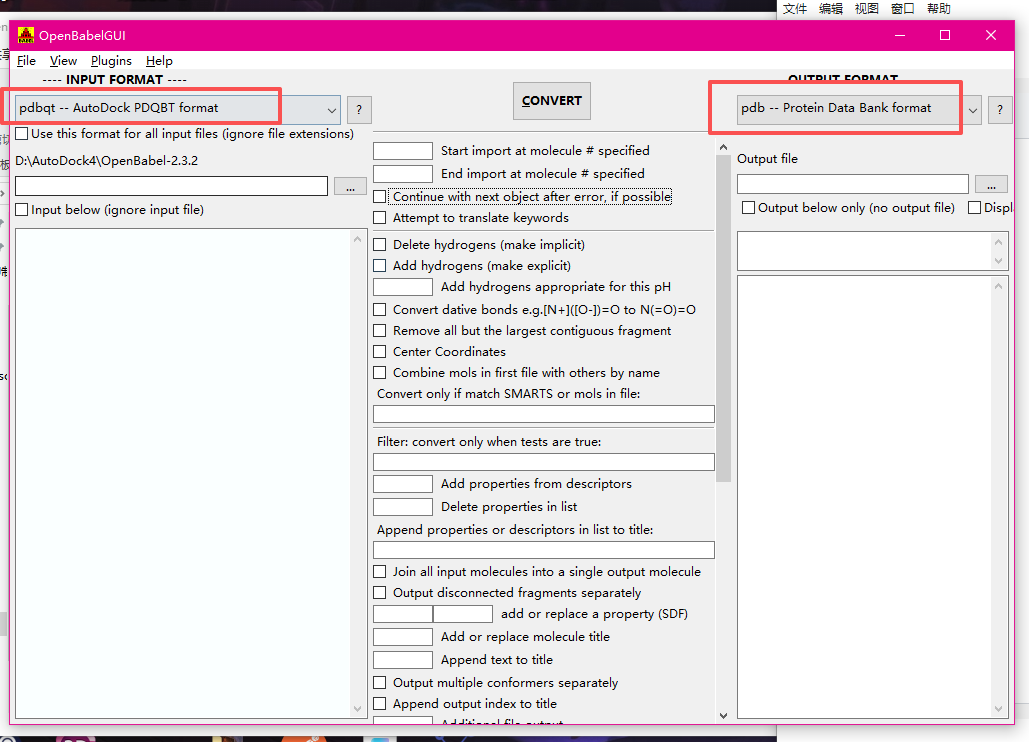
先把输入输出进行调整
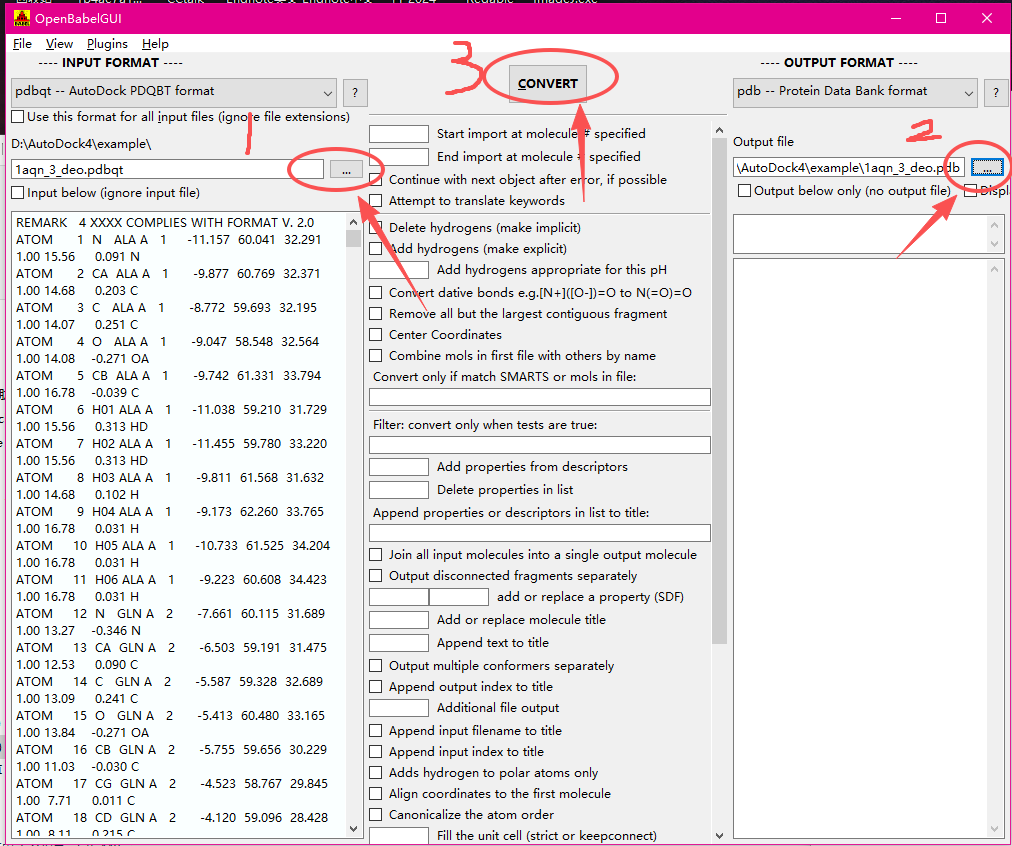
导入文件,然后点击转换
转换完全后,用pymol打开即可
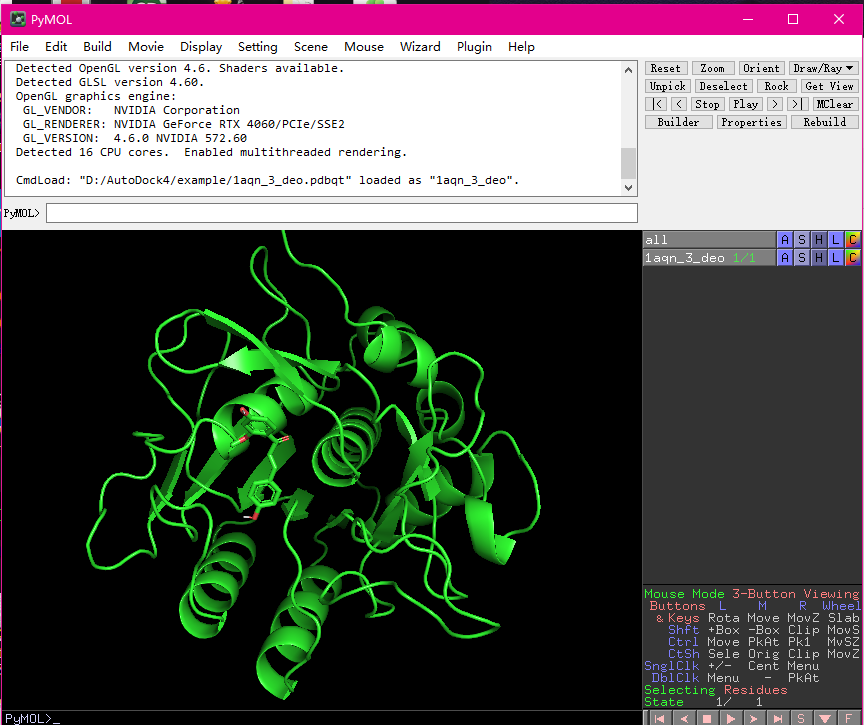
这个就是我们所需要的对接结果