RNA-seq基本介绍及实战——质量控制
实战环节
上一期对转录组学做了一个基本介绍,现在开始进入实战环节
数据预处理
数据预处理在做之前,需要作准备工作,而准备工作一般是准备以下工作
准备工作
- 构建项目目录
- 参考序列下载
- 原始数据上传
构建项目目录
进行转录组分析所使用的平台一般是linux系统,一个常见的工作目录结构如下:
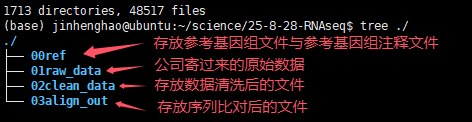
参考序列下载
参考序列一般来说我们需要两个文件
- 参考基因组(fasta格式)
- 注释信息 (gtf/gff格式)
参考序列可以在ensemble数据库获得
里面包含了人类,小鼠等基因组的数据;
另外可访问JGI数据库

本次实例所用的数据库为TAIR数据库、对拟南芥基因库进行下载。
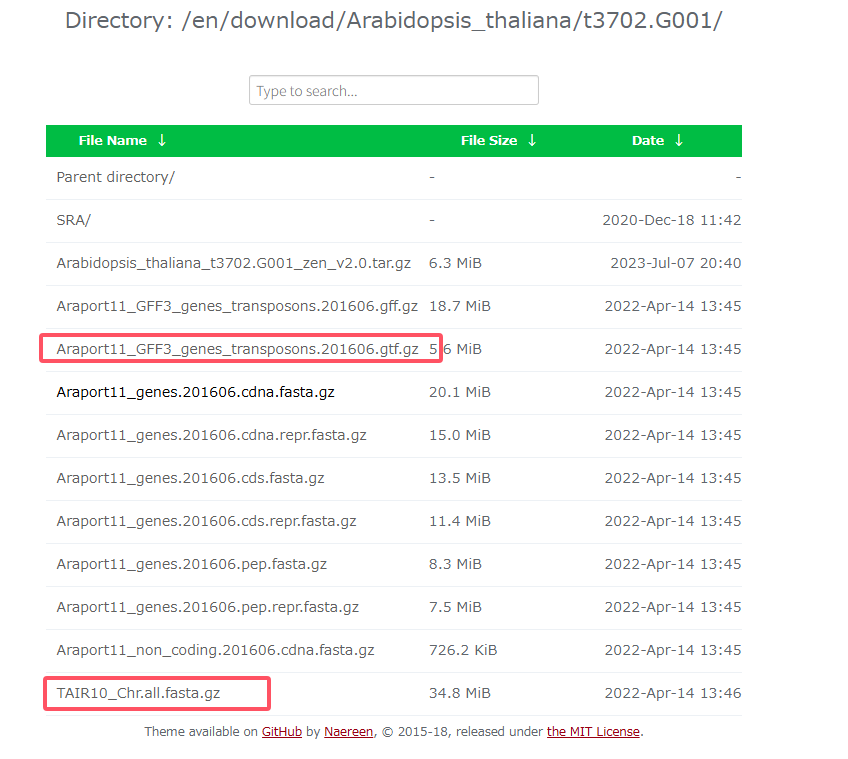
进入00ref目录,用wget命令进行下载
1 | wget https://plantgarden.jp/en/download/Arabidopsis_thaliana/t3702.G001/Araport11_GFF3_genes_transposons.201606.gtf.gz |
当然也可以用curl命令进行下载,这里不作赘述
下一步进行解压
1 | gunzip *gz |
在准备工作完成后,下一步就开始做数据预处理。首先需要做数据的质量控制。一般我们做QC用到的主要有两个:
- FastQC
- MutiQC
这里我们首先用fastqc进行我们的工作
首先需要安装fastaqc,对于linux系统的用户来说,有三种安装方法:
1 | #通过conda安装 |
下一步开始对样本进行qc,输入以下命令:
1 | # 单个样本的qc |
进行qc操作后,此时的文件有以下这些:
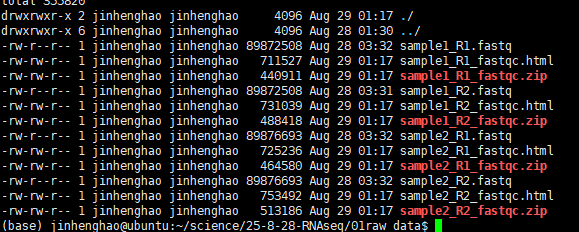
我们把fastqc的结果拿出来拖入浏览器查看一下:
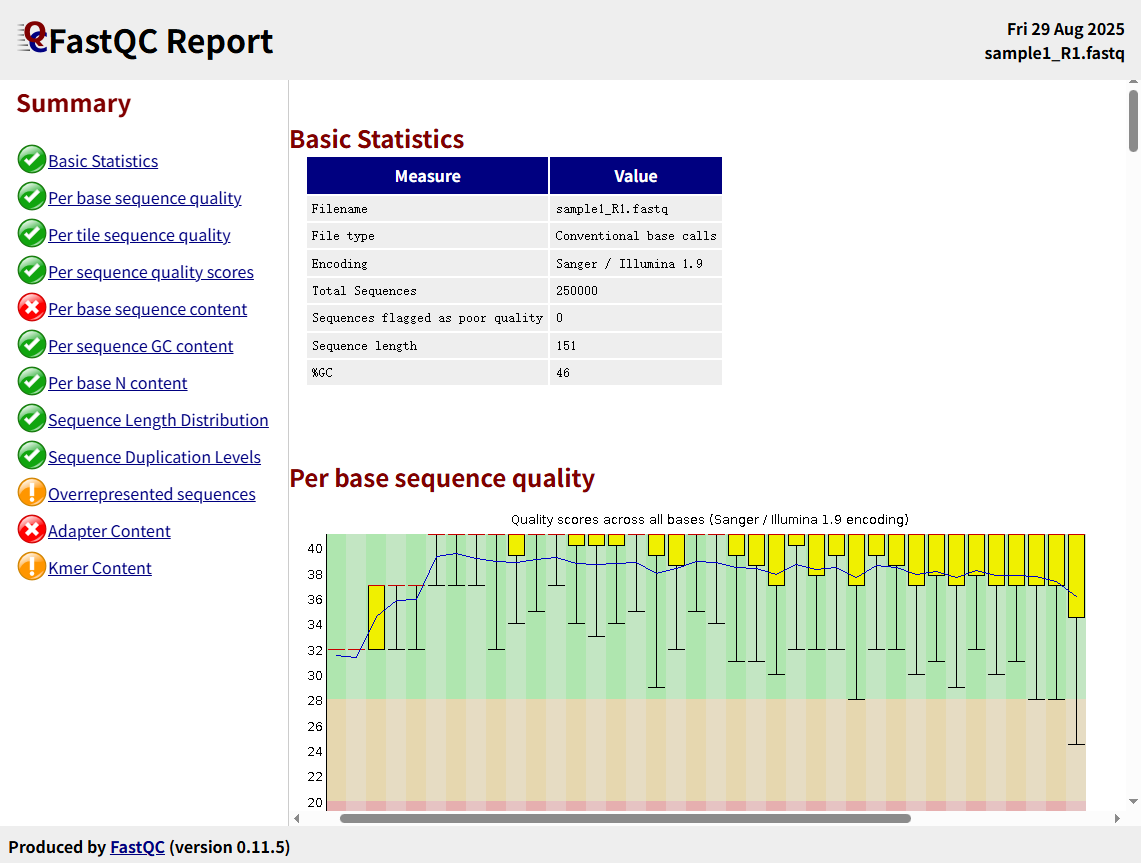
- Per base sequence quality
正常的结果碱基质量全在 Q30 以上,而质量低的结果包含大量 G30 以下的序列,且质量随着读长增加而下降
- Per tile sequence quality
横轴表示碱基位置,纵轴表示 tile 的 index 编号,蓝色表示测序质量很高,暖色表示测序质量不高。tile:每一次测序荧光扫描的最小单位。纵轴是 tail 的 Index 编号。检查 reads 中每一个碱基位置在不同的测序小孔之间的偏离度,蓝色表示低于平均偏离度,偏离度小,质量好;越红表示偏离平均质量越多,质量也越差。如果出现质量问题可能是短暂的,如有气泡产生,也可能是长期的,如在某一小孔中存在残骸,问题不大。
- Per sequence quality scores
每个 reads 的平均质量分布,可以看出低质量结果会在 Q30 以下的部位出现一些杂峰,而高质量结果的峰值在 Q30 以上且无杂峰
后面的结果不再阐述,具体可参考:https://zhuanlan.zhihu.com/p/19856288800
当然fastqc一次也只能分析出一个结果,如果我们有大量的样品的话,做多样品的质量控制,要一个个点开去查看,就显得非常麻烦,这时候我们可以使用multqc进行质量控制。
multqc的安装同样也很简单
1 | # pip安装 |
multiqc的批量化运行也很简单,只需要在放入fastaq文件的目录内执行:
1 | multiqc ./ |
即可运行
运行后结果如此,我们把report文件拿出来看一下:
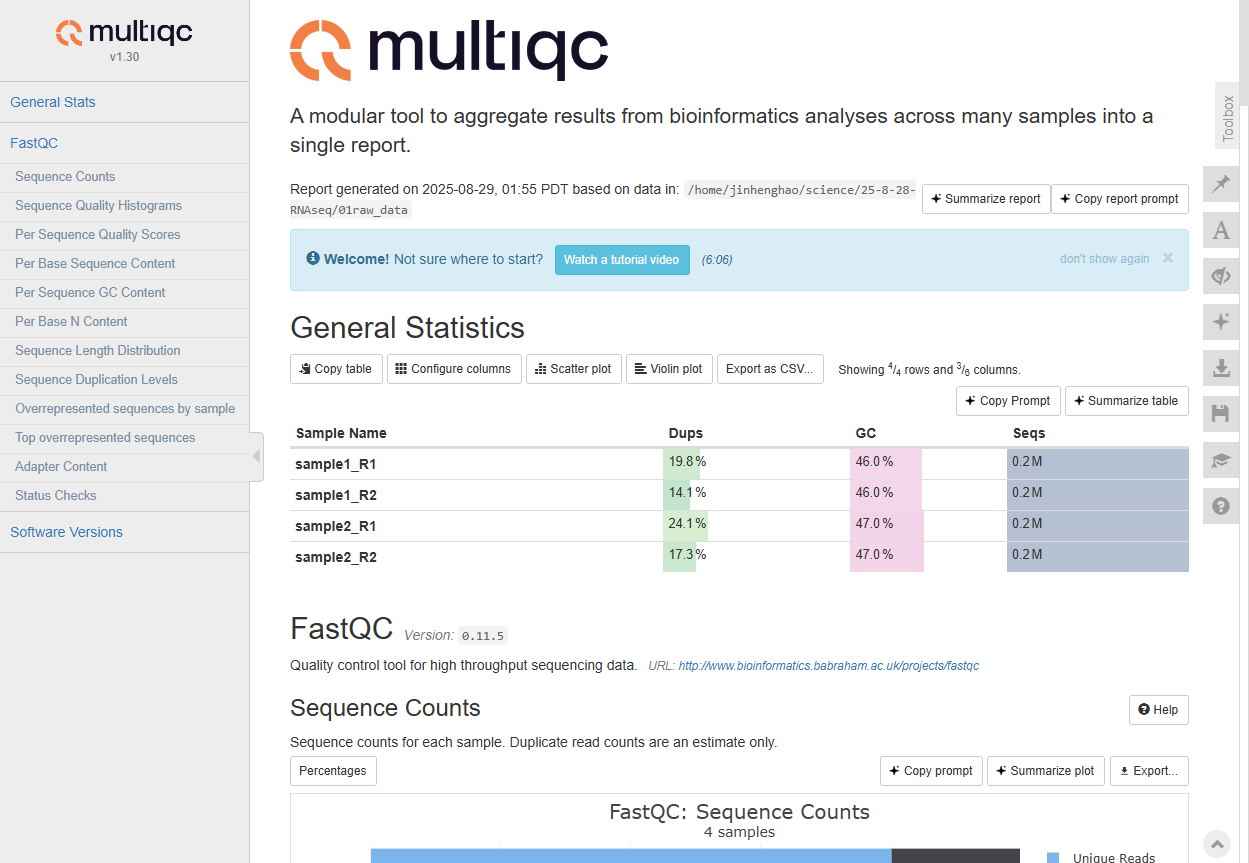
其结果的解读与fastqc差不多相同,这里不作阐述
在进行质量控制之后,我们就需要进行质量过滤,将不符合要求的数据删除掉。这里我们主要使用的是Trimmomatic这个软件,它有以下特点:
- 支持多线程,处理速度快
- 主要用于去除lllumina平台接头
- 根据碱基质量值对fastq进行筛选
- 支持SE和PE测序数据,支持gzip和bzip2压缩文件
Trimmomatic可以用来处理 Illumina (FASTQ)数据以及删除adapter。程序有两种主要模式:Paired-End 模式和 Single-End 模式。Paired-End 模式将保持 read-pairs 的对应关系,并利用 read-pairs 所包含的附加信息,更好地找到文库制备过程中引入的 adapter 或 PCR 引物片段。支持使用 gzip 或 bzip2 压缩的文件,并通过使用.gz 或.bz2 文件扩展名来识别。
目前的 Trimmomatic 主要包含下面步骤
- ILLUMINACLIP: 从 read 中切除 adapter 和 Illumina 的特殊序列。
- SLIDINGWINDOW: 从 read 的 5’端开始滑动窗口,当窗口内的 average quality 小于阈值时,切除后边的碱基.。
- MAXINFO: 自适应的 trim 策略,可以平衡 read 的长度和错误率保证 read 的长度最长。
- LEADING: 切除 read 的开头质量低于阈值的碱基。
- TRAILING: 切除 read 的末尾质量低于阈值的碱基。
- CROP: 从 read 的末尾切除特定长度。
- HEADCROP: 从 read 的开头切除特定长度。
- MINLEN: 舍弃小于最小长度的 reads 。
- AVGQUAL: 舍弃平均质量 小于阈值的 reads。
- TOPHRED33: 将质量分数转换为 Phred-33
- TOPHRED64: 将质量分数转换为 Phred-64
该软件的安装同样也非常简单:
1 | # conda安装 |
使用trimmomatic之前需要下载安装java,其使用方法为:
1 | $ java -jar Trimmomatic-0.39/trimmomatic-0.39.jar -h |
为了简便操作,在这里设置了环境变量。使得单独使用trimmomatic时,也能跳出命令:
1 | sudo vim ~/.bashrc |
而后,输入以下命令开始进行过滤,定位在/01raw_data目录下运行:
1 | trimmomatic PE -threads 8 sample1_R1.fastq sample1_R2.fastq ../02clean_data/sample1_paired_clean_R1.fastq ../02clean_data/sample1_unpaired_clean_R1.fastq ../02clean_data/sample1_paired_clean_R2.fastq ../02clean_data/sample1_unpaired_clean_R2.fastq ILLUMINACLIP:/home/jinhenghao/science/trimmomatic/Trimmomatic-0.39/adapters/TruSeq3-PE-2.fa:2:30:10:1:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:50 TOPHRED33 |
接下来解释下这个命令的意思:
- threads:运行该软件时用到多少线程
- 输入文件:sample1_R1.fastq、sample1_R2.fastq
- 输出文件:sample1_paired_clean_R1.fastq、 sample1_paired_clean_R2.fastq (保存成对信息)sample1_unpaired_clean_R2.fastq、sample1_unpaired_clean_R1.fastq (非成对信息)
- ILLUMINACLIP:/home/jinhenghao/science/trimmomatic/Trimmomatic-0.39/adapters/TruSeq3-PE-2.fa:2:30:10:1:true :这里表示的是调用某文件去掉接头序列,位置入上面路径所示。
做到这里,质控这个过程就结束了,下一步就需要做序列比对。在下一期实战中会进行阐述