rsem-extract-reference-transcripts arab_RSEM/arab_rsem 0 00ref/Araport11_GFF3_genes_transposons.201606.gtf None 0 00ref/TAIR10_Chr.all.fasta Parsed 200000 lines Parsed 400000 lines Parsed 600000 lines Parsed 800000 lines Parsing gtf File is done! 00ref/TAIR10_Chr.all.fasta is processed! 58699 transcripts are extracted. Extracting sequences is done! Group File is generated! Transcript Information File is generated! Chromosome List File is generated! Extracted Sequences File is generated!
rsem-preref arab_RSEM/arab_rsem.transcripts.fa 1 arab_RSEM/arab_rsem Refs.makeRefs finished! Refs.saveRefs finished! arab_RSEM/arab_rsem.idx.fa is generated! arab_RSEM/arab_rsem.n2g.idx.fa is generated!
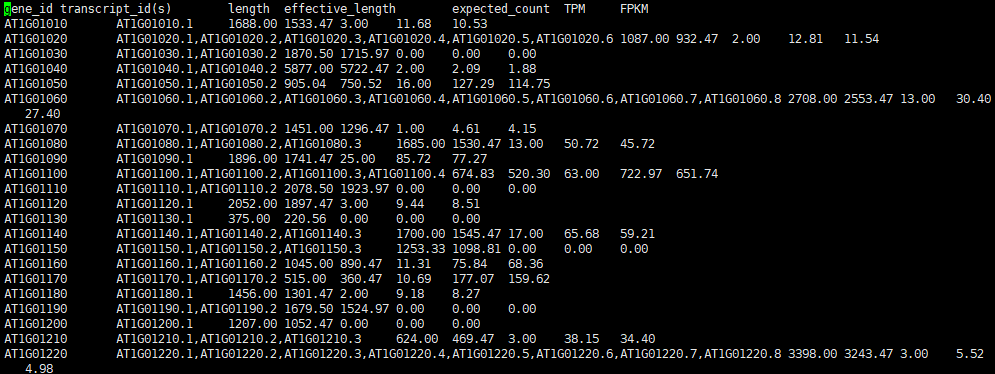
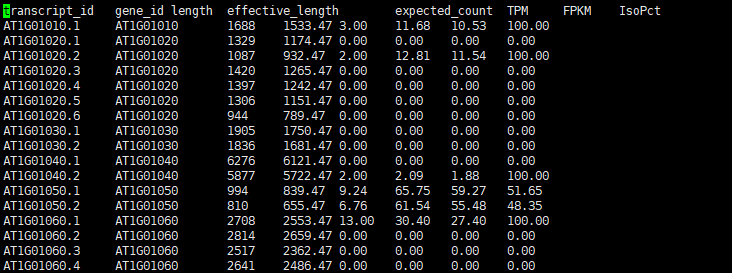
rsem-run-em arab_RSEM/arab_rsem 3 04rsem_out/sample1_rsem 04rsem_out/sample1_rsem.temp/sample1_rsem 04rsem_out/sample1_rsem.stat/sample1_rsem -p 5 -q Time Used for EM.cpp : 0 h 00 m 10 s
[build] loading fasta file ../arab_RSEM/arab_rsem.transcripts.fa [build] k-mer length: 31 [build] warning: clipped off poly-A tail (longer than 10) from 26 target sequences [build] warning: replaced 697 non-ACGUT characters in the input sequence with pseudorandom nucleotides KmerStream::KmerStream(): Start computing k-mer cardinality estimations (1/2) KmerStream::KmerStream(): Start computing k-mer cardinality estimations (1/2) KmerStream::KmerStream(): Finished CompactedDBG::build(): Estimated number of k-mers occurring at least once: 54535532 CompactedDBG::build(): Estimated number of minimizer occurring at least once: 13421357 CompactedDBG::filter(): Processed 97555899 k-mers in 58699 reads CompactedDBG::filter(): Found 54357654 unique k-mers CompactedDBG::filter(): Number of blocks in Bloom filter is 372803 CompactedDBG::construct(): Extract approximate unitigs (1/2) CompactedDBG::construct(): Extract approximate unitigs (2/2) CompactedDBG::construct(): Closed all input files
CompactedDBG::construct(): Joining unitigs CompactedDBG::construct(): After join: 319356 unitigs CompactedDBG::construct(): Joined 24543 unitigs [build] building MPHF [build] creating equivalence classes ... [build] target de Bruijn graph has k-mer length 31 and minimizer length 23 [build] target de Bruijn graph has 319356 contigs and contains 54404035 k-mers
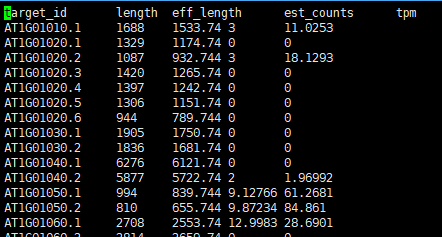
[quant] fragment length distribution will be estimated from the data [index] k-mer length: 31 [index] number of targets: 58,699 [index] number of k-mers: 54,404,035 [quant] running in paired-end mode [quant] will process pair 1: 02clean_data/sample1_paired_clean_R1.fastq 02clean_data/sample1_paired_clean_R2.fastq [quant] finding pseudoalignments for the reads ... done [quant] processed 196,007 reads, 194,599 reads pseudoaligned [quant] estimated average fragment length: 155.256 [ em] quantifying the abundances ... done [ em] the Expectation-Maximization algorithm ran for 766 rounds